
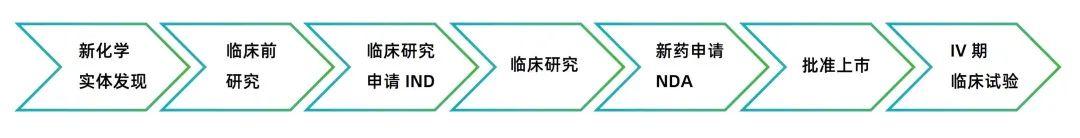
In the field of medicine, the research and development and marketing of new drugs is a rigorous and complex process, involving many links and strict supervision. The drug approval process of the U.S. Food and Drug Administration (FDA) has an important influence worldwide, and the Investigational New Drug (IND) application is a crucial step in this process. It not only concerns whether pharmaceutical companies can smoothly promote the clinical trials of new drugs, but also serves as an important checkpoint to ensure the safety and effectiveness of drugs. The following will detail the Investigational New Drug (IND) application under the FDA drug approval process.
Current Federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across state lines. Because a sponsor will probably want to ship the investigational drug to clinical investigators in many states, it must seek an exemption from that legal requirement. The IND is the means through which the sponsor technically obtains this exemption from the FDA.
Simply and popularly speaking, the IND application is a stage in the new drug approval process of the U.S. Food and Drug Administration (FDA). When a pharmaceutical company develops a new drug, it needs to submit an IND application to the FDA before conducting large-scale clinical trials. Once the IND application is approved, it means that the drug can begin clinical trials on humans. For example, a pharmaceutical company has developed a new targeted drug for cancer. After completing the preliminary cell experiments and animal experiments and believing that the drug has certain therapeutic prospects, it will prepare the IND application materials and submit them to the FDA. If approved, it can recruit patients to conduct Phase I clinical trials to observe the safety, tolerability, pharmacokinetics and other indicators of the drug in the human body.
There are three IND types:
An Investigator IND is submitted by a physician who both initiates and conducts an investigation, and under whose immediate direction the investigational drug is administered or dispensed. A physician might submit a research IND to propose studying an unapproved drug, or an approved product for a new indication or in a new patient population.
Emergency Use IND allows the FDA to authorize use of an experimental drug in an emergency situation that does not allow time for submission of an IND in accordance with 21CFR , Sec. 312.23 or Sec. 312.20. It is also used for patients who do not meet the criteria of an existing study protocol, or if an approved study protocol does not exist.
Treatment IND is submitted for experimental drugs showing promise in clinical testing for serious or immediately life-threatening conditions while the final clinical work is conducted and the FDA review takes place.
The IND application must contain information in three broad areas:
Animal Pharmacology and Toxicology Studies - Preclinical data to permit an assessment as to whether the product is reasonably safe for initial testing in humans. Also included are any previous experience with the drug in humans (often foreign use).
Manufacturing Information - Information pertaining to the composition, manufacturer, stability, and controls used for manufacturing the drug substance and the drug product. This information is assessed to ensure that the company can adequately produce and supply consistent batches of the drug.

Clinical Protocols and Investigator Information - Detailed protocols for proposed clinical studies to assess whether the initial-phase trials will expose subjects to unnecessary risks. Also, information on the qualifications of clinical investigators--professionals (generally physicians) who oversee the administration of the experimental compound--to assess whether they are qualified to fulfill their clinical trial duties. Finally, commitments to obtain informed consent from the research subjects, to obtain review of the study by an institutional review board (IRB), and to adhere to the investigational new drug regulations.

Once the IND is submitted, the sponsor must wait 30 calendar days before initiating any clinical trials. During this time, FDA has an opportunity to review the IND for safety to assure that research subjects will not be subjected to unreasonable risk.
The term "pharmaceutical companies going global" is a hot topic, but at the same time, it faces more and more challenges. Regulatory policies, cultural differences, and language barriers in different countries and regions may all become stumbling blocks for pharmaceutical companies to expand the international market. Language services play an indispensable role in this process. Accurate translation and professional localization services can ensure the integrity and compliance of pharmaceutical companies' application materials and avoid misunderstandings or delays caused by language problems. Glodom, as a language service provider, has rich experience in the field of life sciences. We have a translation team proficient in pharmaceutical expertise and are familiar with the regulatory terms related to drug approval in various countries, providing pharmaceutical companies with accurate and high-quality translation services. We are committed to helping pharmaceutical companies go global and providing professional language services on the way.
The article's information is sourced from CDER and compiled by Glodom.
